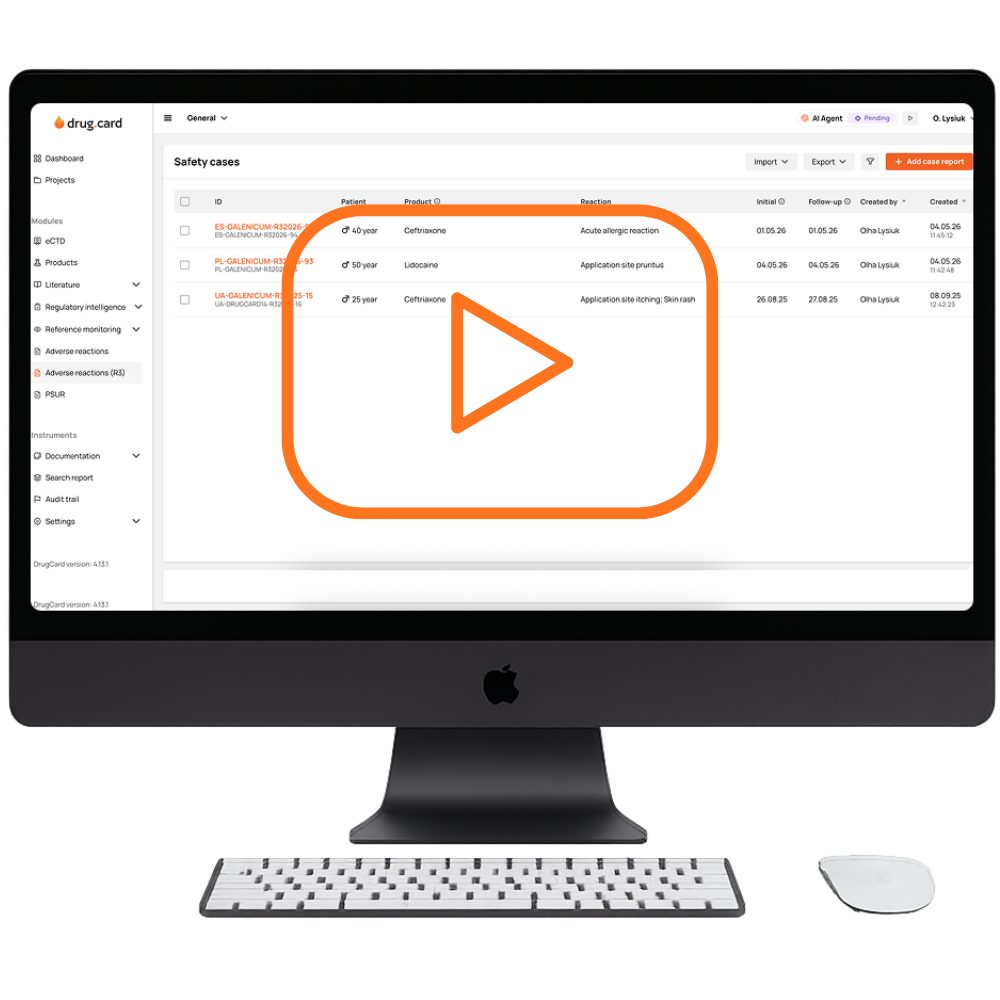
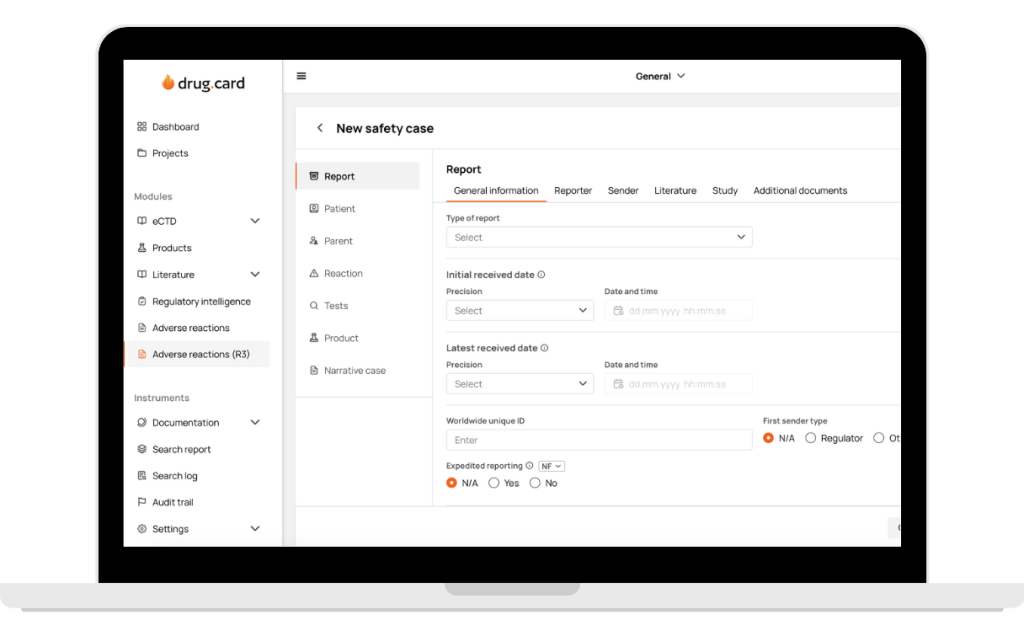
A fully protected adverse event reporting environment is hosted on ISO/IEC 27001:2013 and SOC 2 Type II–certified data centers, featuring built-in GxP-aligned controls such as user access roles, audit trails, and E2B(R2/R3) XML handling, and ensuring full compliance with industry standards.